




Long QT syndrome (LQTS) is a hereditary disorder characterised by prolongation of the QT interval on electrocardiogram (ECG), syncope and sudden death due to torsade de pointes (Tdp) and/ or ventricular fibrillation.1 To date, 16 genes have been reported as LQTS-causing genes, and most mutations identified in LQTS patients are in the KCNQ1, KCNH2 and SCN5A genes, which correspond to types 1, 2, and 3 LQTS (LQT1, LQT2 and LQT3), respectively. Implantable cardioverterdefibrillator (ICD) implantation, beta-blocker therapy and left cardiac sympathetic denervation are recommended for the prevention of sudden death in this patient population.1 The classic ‘R-on-T’ phenomenon is a well-known entity;2 however, the ‘T-on-P’ phenomenon is rarely reported in literature. We present an asymptomatic two-month-old infant with innocent murmur and ECG showing a corrected QT interval of 600 msec, who underwent ICD placement at our centre. The genetic analysis revealed a homozygous mutation in the KCNQ1 gene (LQT1).
Case report
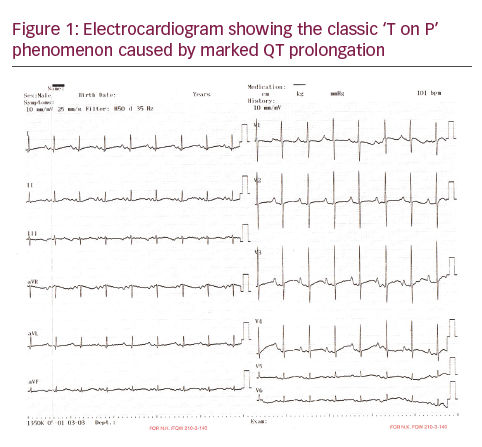
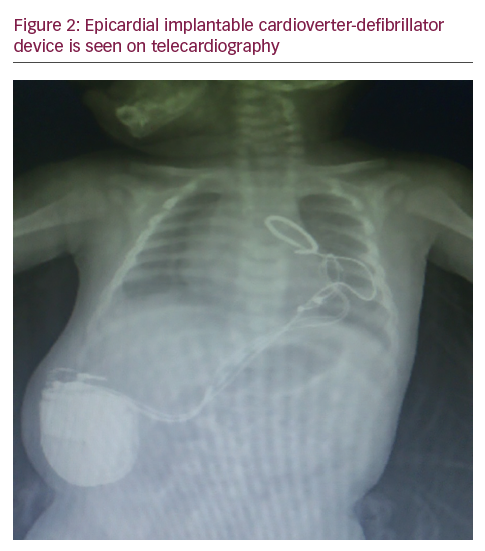
A two-month-old infant referred to our hospital for innocent murmur. His parents had no complaints about their baby. Examination was normal except innocent murmur and echocardiography was unremarkable, which showed the prolonged corrected QT interval of 600 msec (see Figure 1A). Twenty-four hour Holter ECG was normal except prolongation of the corrected QT (QTc) interval with typical ‘T on P’ phenomenon (see Figure 1). The genetic analysis revealed a homozygous p.R366Q (c.1097>A) mutation in the KCNQ1 gene (LQT1). The hearing test was unrevealing. Although seven brothers of the patient died before reaching age one year with no known reason, the QT/QTc interval of his father and brother were in the normal range in addition to a normal QTc interval of his mother. Oral beta-blocker (propranolol) (3 mg/kg/day) was administered and ICD implantation was planned. Under general anaesthesia, a short right lateral paraumblical incision was made for the ICD implantation. Using the previous subxhyphoid incision from the pacemaker, both electrodes of the 35 cm bipolar, steroid-eluting, pace-sensing lead (Bipolar CapsureEpi IS-1, Model 4968, Medtronic; Minneapolis, Minnesota, US) were attached to the epicardium of the anterior right ventricular wall. A subcutaneous defibrillation electrode (35 cm, unipolar endocardial superior vena cava lead for cardioversion and defibrillation; Unipolar Transvene-SVC Model 6937, Medtronic) was passed through the right rectus muscle and tunnelled subcutaneously to a position below the left scapula. Subsequently, the ICD device (Maximo II VR D284VRC, Medtronic) was inserted into an abdominal pocket behind the right rectus abdominis muscle (see Figure 2). The boy’s post-operative course was uneventful, and he was discharged 3 days after the operation with propranolol 3 mg/kg/day. During the six-month follow-up period, the boy had no attacks of ventricular tachycardia or ventricular fibrillation.
Discussion
In this article, we presented an ECG finding with classic ‘T on P’ phenomenon consisting of prolongation of Q and a delayed T wave, followed or overlapped by the succeeding P wave.3 It has been demonstrated that congenital LQTS with very long QT intervals can produce a functional block between the His bundle and ventricular muscle due to prolonged ventricular refractoriness that can lead to 2:1 atrioventricular block and severe bradycardia.3
Genetic testing not only has significant diagnostic implications but also has prognostic and therapeutic implications in LQTS. For example, the underlying genetic basis heavily influences the response to standard LQTS pharmacotherapy (beta-blockers), because beta-blockers are extremely protective in LQT1 patients but not as protective for patients with either LQT2 or LQT3.4 In addition, intra genotype risk stratification has been realised for LQT1 and LQT2 on the basis of mutation type, mutation location and cellular function.4 Therefore, genetic analysis of LQTS is important to identify the therapeutic option and risk stratification.5 In our patient, the genetic analysis identified a missense mutation (R366Q) in the KCNQ1 gene. Propranolol therapy was useful in this patient because beta-blockers are extremely protective in LQTS1 patients. In the current patient, additional treatment with
ICD was performed because the QTc interval was very long at 600 msn and male sex.
Implantation of ICDs in paediatric patients presents various difficulties.6 Despite the downsizing of pulse generators as well as improvements in lead design, paediatric patients present challenges in terms of patient size and growth. Relative to the venous diameter and thoracic anatomy of the infants, transvenous leads are large in calibre and length. Epicardial patches require sternotomy or thoracotomy, with higher rates of insulation and conductor fractures as well as the risk of the development of restrictive pericardial lesions due to the large surface area of the patches. Implantation techniques using a subcutaneous array and an abdominally placed ICD generator have been described previously.6 However, experience with infants younger than six months is limited. Despite low body weight and young age, ICD implantation applied to this child as a primary prophylaxis in LQTS. ICD implantation is an effective therapy and can be applied to small children successfully.
