Hypertrophic cardiomyopathy (HCM), along with its more common subgroup, hypertrophic obstructive cardiomyopathy (HOCM), is the most common monogenic cardiovascular disorder, with a prevalence of about 1 in 500,1 and a dominant inheritance pattern, thus clustering in affected families. Patients with HCM often have significant limitations to their exercise capacity, and while there are several underlying pathomechanisms for common symptoms, such as dyspnoea, angina and fatigue, one major cause and treatment target is left ventricular outflow tract (LVOT) obstruction.2,3 An increased LVOT gradient (i.e. the quantitative measure of obstruction) occurs due to an interplay between several contributors: (1) left ventricular septal hypertrophy leading to a narrow LVOT, (2) systolic anterior motion of the anterior mitral leaflet from either a Venturi (i.e. suctioning) effect or a posterior-to-anterior systolic flow direction causing a push of the mitral leaflets into the LVOT, (3) abnormal papillary muscle configuration, (4) excessive anterior mitral leaflet tissue and (5) a small left ventricular cavity with hyperdynamic contractility. The latter mechanism has been the primary target for pharmacological therapies to reduce the LVOT gradient.4 However, until the discovery of myosin adenosine triphosphate (ATP) inhibitors, the effectiveness of available drug therapies was suboptimal in many patients and was often based on small or non-randomized studies.
Myosin ATP inhibitors are a new drug class targeting one underlying cause of the development of hypertrophy, inefficient cellular energetics and LVOT obstruction: excessive actin–myosin cross-bridging.5–9 The reduction of actin–myosin interactions decreases contractile force, thereby reducing LVOT gradient. However, there is preliminary evidence of mavacamten also having potential beneficial effects on diastolic function10 and microvascular blood flow and improving in cardiac structure.11 In this article, we review the preclinical and clinical data that make this novel drug class an attractive new option for the management of patients with symptomatic HOCM
The hypertrophic cardiomyopathy disease process
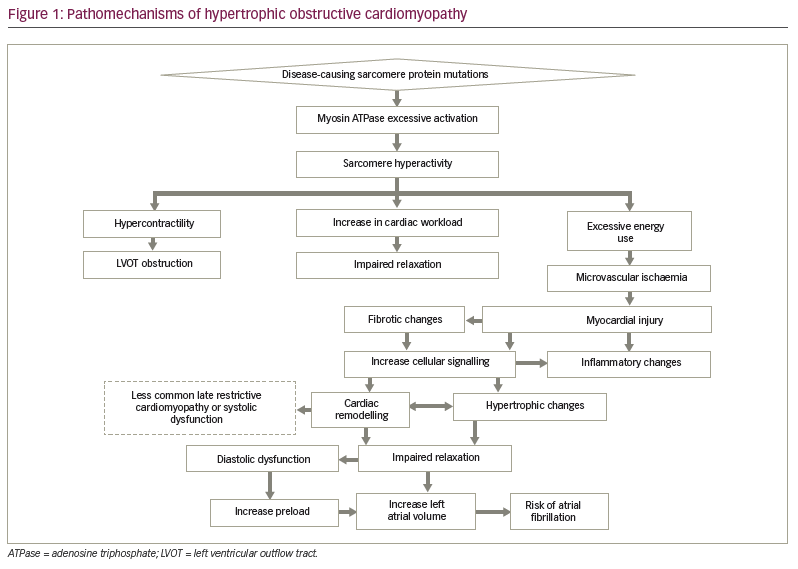
The pathophysiology of HCM is not yet fully understood, as it is complex and multifactorial.12 However, the most accepted pathomechanism is that HCM is a disease process caused by sarcomeric dysfunction secondary to missense mutations of one or several genes coding for contractile proteins, mainly the beta-myosin heavy chain, myosin-binding protein C or troponin.13,14 These genetic alterations lead to changes in sarcomere physiology, such as hypersensitivity of myofilaments to calcium and an increase in myosin adenosine triphosphatase (ATPase) activity. These changes in sarcomere physiology promote the shifting of more myosin heads from the physiologically relaxed state to the active state.6 In a normal cardiomyocyte, 40–50% of the myosin heads are in the relaxed state, resulting in minimal energy consumption.15–17 In contrast, HCM-afflicted cardiomyocytes only have 15–20% of myosin heads in the relaxed state, which results in excessive activation and unnecessary energy consumption.15–17 Excess activation of myosin heads not only consumes energy in the form of ATP through the inappropriate activation of myosin ATPase but also predisposes the myosin heads to bind actin more readily, resulting in an increase of actin–myosin cross-bridging throughout the cardiac cycle.6,18–21 Sarcomere hyperactivity leading to myocardial hypercontractility is an important process in the pathophysiology of HCM because it has been linked to an increase in cellular signalling that leads to hypertrophic changes.22,23 Ultimately, this results in detrimental cardiac remodelling brought about by fibrotic change, myofibre hypertrophy with disarray secondary to increased activation of pathways involving hypertrophy, inflammation and fibrosis (Figure 1).18–20 These changes lead to outflow tract obstruction and diastolic dysfunction; they thus lead to an increase in cardiac workload, rendering the heart an ineffective pump, hence leading to the development of symptoms of heart failure (i.e. the inability to maintain normal left atrial pressure).22
Molecular mechanisms of mavacamten
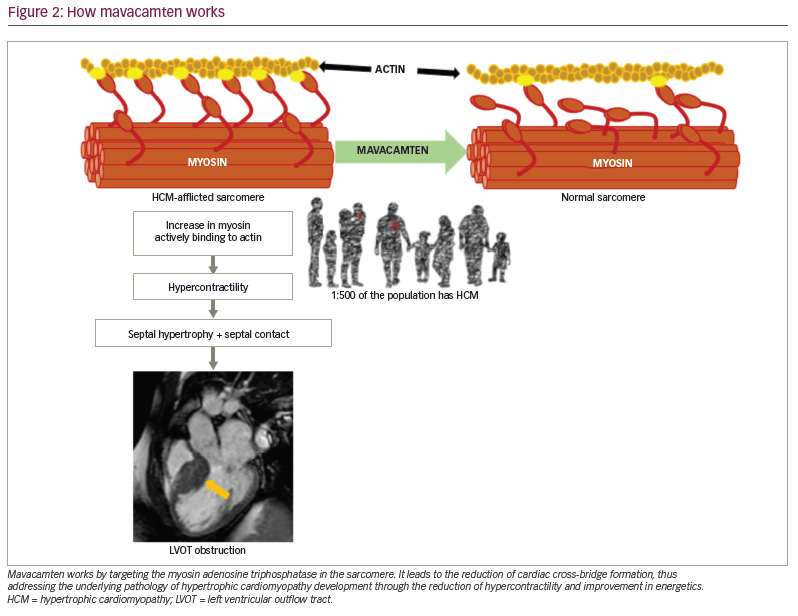
Mavacamten, formerly known as MYK-461, is a first-in-class medication referred to as a cardiac myosin ATPase inhibitor that has recently been approved by the US Food and Drug Administration as a treatment for symptomatic HOCM.9 Mavacamten is a small molecule that acts selectively and reversibly on the allosteric binding site of the cardiac myosin ATPase enzyme (Figure 2).15 Recent studies have shown that the inhibition of cardiac myosin ATPase mitigates the excessive ATP expenditure seen in HCM by preventing the myosin heads from unnecessarily shifting from the relaxed energy-sparing state to the active energy-expending state.17 As a result, mavacamten reduces actin–myosin cross-bridge formation, thus addressing the underlying pathology of HCM development through the reduction of hypercontractility and improvement in energetics.5–8 Furthermore, these molecular changes from mavacamten optimize the mechanical environment of the sarcomere by reducing diastolic tension with improved relaxation, increasing ventricular chamber size, reducing myocardial contraction velocity and ultimately decreasing outflow obstruction.5,17,20,23
The primary focus in hypertrophic obstructive cardiomyopathy: The left ventricular outflow tract gradient
HOCM is the most prevalent subgroup of HCM, making up approximately 70% of HCM occurrences; Its definying features include asymmetric ventricular septal hypertrophy and systolic anterior motion of the mitral valve.2 The presence of LVOT obstruction at rest, defined as an LVOT gradient of ≥30 mm, is associated with a twofold increase in the risk of HCM-related death,3 suggesting that obstruction in HCM is not only causing symptoms but is also a treatment target for improving patients’ longevity. LVOT obstruction is present in about one-third of patients at rest, and another 40% of patients develop LVOT obstruction during exercise or other provocative manoeuvres, which therefore are essential in the assessment of HCM patients.4 Patients with LVOT obstruction also have a greater risk of developing clinical symptoms, arrhythmias and other HCM-related complications.1
The available pharmacotherapy (i.e. the initial and preferred treatment approach) has been limited; although it is highly effective in some, it has left others with significant residual symptoms that are attributable, at least in part, to residual LVOT obstruction.24 Before the availability of mavacamten, treatment options were non-dihydropyridine calcium channel blockers, non-vasodilating beta-blockers and disopyramide, which mainly work by modestly decreasing contractility and allowing for prolonged ventricular filling time, thereby increasing left ventricular (including LVOT) size.25–28 In people who have severe symptoms from persistent obstruction despite maximal-tolerated medical treatment, invasive septal reduction therapy can and should be considered; however, although effective, this therapy has an indisputably small but not negligible risk of complications29,30 and in some cases does not provide a complete resolution of obstruction and associated symptoms.31 In addition, neither myectomy nor alcohol septal ablation completely abolishes concomitant myocardial abnormalities typical in HCM, such as abnormal wall tension, diastolic dysfunction and microvascular ischaemia, from poor myocardial energetics.
Preclinical studies of mavacamten
In a preclinical study, the administration of mavacamten prevented the development of hypertrophy in mice with HCM who had yet to develop myocardial thickening and reversed hypertrophy in mice with HCM with established hypertrophic cardiac changes due to a reduction in the expression of profibrotic and pro-hypertrophic genes.32 Histopathological studies confirmed that mavacamten prevented the development of myocardial fibrosis and cardiomyocyte disarray. However, some of these changes remained irreversible, as confirmed by histopathology showing persistent myocardial fibrosis and cardiomyocyte disarray.32
Furthermore, the histopathology of untreated hearts of mice with HCM showed patchy fibrosis throughout the myocardium by Masson’s trichrome staining, similarly to what can be seen in hearts of humans with HCM. Conversely, mice with HCM treated with mavacamten had minimal fibrosis.32
Mavacamten led to a dose-dependent decrease in maximal tension and fractional shortening in an isolated cardiac muscle fibre model.31 In addition, mavacamten led to regression of left ventricular wall thickness in mice with HCM.32
Taken together, these studies in mice with HCM suggest that mavacamten not only has the potential to induce regression of hypertrophy but may also prevent the development of cardiac hypertrophy and fibrosis. These findings indicate that mavacamten may show a similar effect in humans. Such early treatment prevention studies have not been performed in humans to date and, unfortunately, would be difficult to carry out.
Clinical studies of mavacamten in patients with hypertrophic cardiomyopathy
PIONEER-HCM
The initial data on the safety and efficacy of mavacamten were obtained from the PIONEER-HCM study, a phase II, open-label, non-randomized study of patients with symptomatic HOCM (A phase 2 open-label pilot study evaluating MYK-461 in subjects with symptomatic hypertrophic cardiomyopathy and left ventricular outflow tract obstruction [PIONEER-HCM]; ClinicalTrials.gov identifier: NCT02842242) (Table 1).33 Key inclusion criteria were age 18–70 years, a clinical diagnosis of HCM, a screening of left ventricular ejection fraction (LVEF) of ≥55% by echocardiography, usual definitions of HCM by maximum left ventricular wall thickness and LVOT obstruction of ≥30 mmHg at rest or ≥50 mmHg post-exercise. What all mavacamten studies have in common are New York Heart Association (NYHA) class II or III symptoms. Patients who had exertional syncope in the past 6 months, sustained ventricular tachycardia, a history of LVEF of <45%, persistent atrial fibrillation, atrial fibrillation with a heart rate of >100 bpm within a year of screening or obstructive coronary artery disease were excluded from the study.33
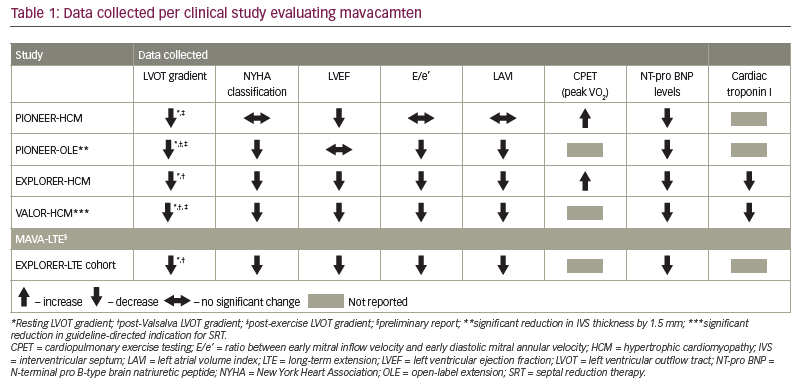
Of 25 participants initially enrolled, 21 completed the study and were assigned either to cohort A (n=11) or cohort B (n=10). Patients in cohort A were started on 10 mg/day if their weight was ≤60 kg or on 15 mg if their weight was >60 kg. Cessation of calcium channel blockers, beta-blockers and disopyramide 14 days prior to the first dose of mavacamten was required in participants of cohort A. Drug titrations were based on changes in LVEF during weekly follow-ups to a dose of up to 20 mg/day. Patients assigned to cohort B were started on lower doses, starting at 2 mg/day with a continuation of beta-blockers at a stable dose. Drug adjustment for cohort B was based on the patient’s LVOT gradient at the end of the week 4: a 5 mg/day increase was initiated if the gradient had decreased by <50% from baseline. At week 12, all patients in cohort B received the 5 mg/day dose increase.33
Patients from cohort A who received a mean mavacamten dose of 15 mg/day demonstrated overall beneficial clinical effects at 12 weeks, with a more profound effect seen among patients on a higher dose. At 12 weeks, the mean post-exercise LVOT gradient decreased from 103 mmHg at baseline to 19 mmHg in cohort A and from 86 mmHg at baseline to 64 mmHg in cohort B. Valsalva LVOT gradient decreased by an average of 85 mmHg (95% confidence interval [CI]: 56–114 mmHg) in cohort A and 47 mmHg (95% CI: 12–82 mmHg) in cohort B. LVEF decreased by 15% (95% CI 6–23%) in cohort A and 6% (95% CI: 1–10%) in cohort B. These data confirm that the effects of mavacamten on both LVOT gradient and LVEF reduction are directly proportional to drug dosage.33
Mavacamten use was also associated with improvement in patients’ functional status, as demonstrated by an average of 0.9 (cohort A) and 1.0 (cohort B) reduction in NYHA class. The Kansas City Cardiomyopathy Questionnaire (KCCQ) also demonstrated improvements, from an average score of 65.0 to 14.4 in cohort A and from 61.0 to 16.0 in cohort B.33
In summary, the PIONEER-HCM study showed promising improvements in LVOT gradient and clinical symptoms among enrolled patients with HOCM and formed the basis for subsequent phase III trials.33
PIONEER-OLE
This PIONEER-OLE study (Extension study of mavacamten [MYK-461] in adults with symptomatic obstructive hypertrophic cardiomyopathy previously enrolled in PIONEER; ClinicalTrials.gov identifier: NCT03496168) was designed to examine the long-term safety and clinical symptoms in patients who completed the previous PIONEER-HCM study (Table 1).34 After medication washout, patients were started on a mavacamten dose of 5 mg/day. At 6 weeks, doses could be increased to 10 or 15 mg. Patient assessments were conducted at 4, 6, 8, and 12 weeks initially and at 12-week intervals thereafter. During follow-up visits, LVEF, LVOT gradient, NYHA class, N-terminal pro B-type brain natriuretic peptide (NT-pro BNP), drug concentrations and adverse effects were assessed.
At 48 weeks, mavacamten use was again associated with improvements in LVOT gradient of 14.0 mmHg and 22.4 mmHg at rest and post-Valsalva, respectively. Improvements in diastolic function were also captured objectively through a 9.8 mL/m2 reduction in the left atrial volume index and a 3.4-point reduction in lateral E/e’ (ratio between early mitral inflow velocity and early diastolic mitral annular velocity). Interestingly, there was a statistically significant reduction of 1.5 mm in interventricular septal thickness, representing the first-in-human evidence for hypertrophy regression in patients with HCM treated with mavacamten. The ejection fraction remained normal, with mean values of 67.6% and no reductions to <50% throughout the study. A mean reduction of 472 pg/mL in NT-pro BNP levels was also observed. Three patients were considered NYHA class I, and three patients were considered NYHA class II. The KCCQ was also reported to have improved.34
Regarding drug toxicity, mavacamten was generally well tolerated, with all adverse events described as mild to moderate and self-resolving. No serious or cardiac events were noted in patients taking mavacamten.34 Although the results of this extension study were promising, major limitations were the small sample size (n=12 at the end of the study) and the non-randomized design.34
EXPLORER-HCM
EXPLORER-HCM (Clinical study to evaluate mavacamten [MYK-461] in adults with symptomatic obstructive hypertrophic cardiomyopathy; ClinicalTrials.gov identifier: NCT03470545) was the pivotal phase III, randomized, double-blind, placebo-controlled, multicentre trial of mavacamten in symptomatic HOCM conducted in 13 countries (Table 1).8 Study participants were diagnosed with HOCM according to the usual criteria, with an LVOT gradient of ≥50 mmHg, an LVEF of ≥55%, NYHA class II or III, and age ≥18 years at screening. Key exclusion criteria were a history of syncope and sustained ventricular tachycardia with exercise within 6 months prior to screening. While background therapy with a beta-blocker or calcium channel blocker was allowed, patients could not be on both. Disopyramide was also washed out prior to enrolment. Of the 251 enrolled patients, 123 were randomized to the mavacamten arm and 128 to the placebo arm. Overall, 97.2% of participants completed the study. Results showed that 36.6% of patients on mavacamten achieved the positive clinical response compared with placebo, which demonstrated an increase in peak volume of oxygen consumption (VO2) of ≥1.5 mL/kg/min plus ≥1 NYHA class improvement, or peak VO2 increase by ≥3.0 mL/kg/min with no worsening of NYHA class at 30 weeks. Peak VO2 improved by 1.4 mL/kg/min from baseline to week 30 among patients who received mavacamten but did not change in the placebo group. The KCCQ administered at 30 weeks of treatment revealed a higher score among patients receiving mavacamten. The Hypertrophic Cardiomyopathy Symptoms Questionnaire Shortness-of-Breath administered at 30 weeks of treatment showed a reduced score in patients given mavacamten, indicating that mavacamten relieved frequency and severity of shortness of breath. In addition, profound reductions in NT-pro BNP by 80% were observed in patients treated with mavacamten.
Overall, this study demonstrated that treatment with mavacamten improved functional capacity, symptom status, LVOT gradient and clinically important reductions in biomarkers. In addition, a magnetic resonance imaging substudy reported the following findings, demonstrating that mavacamten was associated with structural improvements: a 17.4 g/m2 reduction in left ventricular mass index and a 10.3 mL/m2 reduction in left atrial volume index.11
In terms of safety and tolerability, patients who were given mavacamten experienced similar adverse events to those experienced in the placebo group; however, there were more reports of dizziness and syncope, which were unlikely to be arrhythmic in nature. One participant in the placebo arm died from sudden death during the study. Overall, seven patients in the mavacamten arm and two patients in the placebo arm developed reductions of LVEF below 50%. In all seven patients, LVEF recovered with cessation of mavacamten; however, this observation led to current requirements for a stringent risk mitigation programme set forth by the US Food and Drug Administration to closely monitor patients for the development of systolic dysfunction and also to avoid prescription of concomitant medications that can affect the metabolism of mavacamten or the metabolism of the concomitant medication (e.g. cytochrome P450 substrates). Six patients, three from each group, underwent temporary discontinuation because they satisfied the predefined criteria for changes in QT interval. There were no temporary discontinuations due to mavacamten plasma concentrations >1,000 ng/mL.8
VALOR-HCM
The VALOR-HCM study was a multicentre, randomized, double-blind, placebo-controlled, phase III study designed to evaluate the safety and efficacy of adding mavacamten to an established medical regimen on reducing the number of people with symptomatic HOCM needing guideline-directed septal reduction therapy after 16 weeks of treatment (A study to evaluate mavacamten in adults with symptomatic obstructive HCM who are eligible for septal reduction therapy; ClinicalTrials.gov identifier: NCT04349072) (Table 1).35
Key inclusion criteria were people with HOCM defined by the usual criteria and an LVOT gradient of ≥50 mmHg at rest or post-exercise who were referred for septal reduction therapy within the last 12 months because of severe exertional symptoms (NYHA classification of III to IV or NYHA classification II if with syncopal episodes) despite maximal medical therapy, which may include disopyramide and/or a combination of beta-blockers and calcium channel blockers. For patients on beta-blockers, calcium channel blockers or disopyramide, doses had to be stable for 14 days.
Out of the 112 participants, 56 patients were randomly assigned to the mavacamten arm and 56 patients to the placebo arm. A starting dose of 5 mg/day of mavacamten was given to each participant in the mavacamten group. These patients were given blinded dose adjustments based on the LVEF and LVOT gradient at rest and with Valsalva provocation.
At 16 weeks, patients were reassessed for eligibility for guideline-directed septal reduction therapy. Only 17.9% of patients who were receiving mavacamten remained eligible for septal reduction therapy compared with 76.8% in the placebo group. The improvement in NYHA class was superior in the mavacamten group compared with the placebo group. Resting LVOT gradients at 16 weeks averaged 14 mmHg in the mavacamten group versus 46 mmHg in the placebo group. The Valsalva LVOT gradient was also notably lower in the mavacamten group than in the placebo group, at 28 mmHg and 78 mmHg, respectively. Patients taking mavacamten scored higher on KCCQ-23, averaging 80 versus 67 in the placebo group, indicating symptom improvement in the former group.
Regarding adverse events, there was no detected non-sustained ventricular tachycardia in the mavacamten group compared with 9.1% in the placebo group. Mild adverse events were higher in patients taking mavacamten in the placebo arm. No patients experienced any serious adverse events; however, temporary drug cessation was necessary for two patients due to LVEF below 50%; no LVEF below 30%, which requires permanent drug cessation, was reported.35
Longer-term effectiveness and safety data: MAVA-LTE
MAVA-LTE is an on-going, dose-blinded extension study (A long-term safety extension study of mavacamten in adults who have completed MAVERICK-HCM or EXPLORER-HCM; ClinicalTrials.gov identifier: NCT03723655) that was conducted on patients who completed either MAVERICK-HCM, a proof-of-concept study in non-obstructive HCM (A phase 2 study of mavacamten in adults with symptomatic non-obstructive hypertrophic cardiomyopathy [nHCM]; ClinicalTrials.gov identifier: NCT03442764) or EXPLORER-HCM (ClinicalTrials.gov identifier: NCT03470545).36 It was designed to assess long-term data in patients with HCM taking mavacamten, including the frequency and severity of treatment-emergent adverse and serious adverse events over 5 years and to demonstrate long-term durability of the initially observed benefits of mavacamten in HCM (Table 1).36
Eligible patients must have completed the parent study, with a persistent LVEF of ≥50% by transthoracic echocardiography assessed by the echo core lab, with laboratory parameters in the normal range for safety.
All patients, principal investigators and site coordinators who perform study assessment are blinded to the mavacamten dose but aware of active treatment. Nine patient assessments are planned in the first year, five in the second year, four in the third year and, finally, two in the fourth and fifth years. Patients undergo transthoracic echocardiography, electrocardiogram, physical examination, blood work, pharmacokinetic studies, accelerometer measurements and implantable cardiac defibrillator checks, if applicable. At the end of each visit, appropriate drug adjustments are recommended by the site investigators for optimal mavacamten dosing (i.e. the lowest effective dose). To date, only interim results from MAVA-LTE have been published; they will likely determine the clinical use of mavacamten in future practice.36,37
Where does mavacamten fit into current hypertrophic cardiomyopathy treatment algorithms?
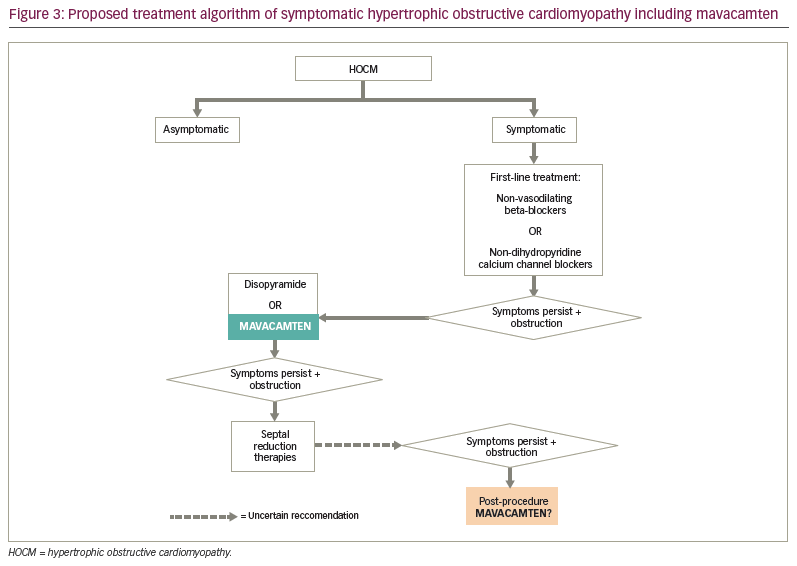
The guiding principle in treating HCM has traditionally been directed towards ameliorating symptoms. Guidelines on the stepwise approach to pharmacological and invasive treatment options are available.28,38–40
Current pharmacological guidelines employ the use of negative chronotropic drugs as initial treatment (non-vasodilating beta-blockers and non-dihydropyridine calcium channel blockers) to decrease heart rate, which in turn prolongs ventricular filling time.38–40 This causes larger left ventricular end-diastolic volume and LVOT with a reduction of obstruction. In addition, the negative inotropic effects of the beta-blockers, the calcium channel blockers and disopyramide reduce contractile forces and LVOT obstruction. Whether beta-blockers and calcium channel blockers worsen or improve diastolic function is incompletely understood. Currently, the data supporting the effect of these pharmacological therapies on the natural history of HCM are incomplete.39–42 There are currently no guideline-recommended algorithms on the optimal use of mavacamten in clinical practice; nonetheless, given some continued access and cost issues and the requirement for frequent and regular clinical and echocardiographic monitoring with mavacamten use, beta-blockers and calcium channel blockers are likely to continue as the first-line treatment option for most patients with HOCM, at least until longer-term data are available (Figure 3). However, such longer-term safety follow-up studies may lessen the clinical monitoring requirements in the future and encourage broader-scale implementation. Additional follow-up studies will also inform the question of whether mavacamten alters long-term clinical outcomes and reverses or halts the development of hypertrophic and fibrotic changes in the affected myocardium. If on-going studies confirm this hypothesis, mavacamten may become the preferred first-line treatment option for symptomatic HOCM in the near future.
Limitations of using mavacamten
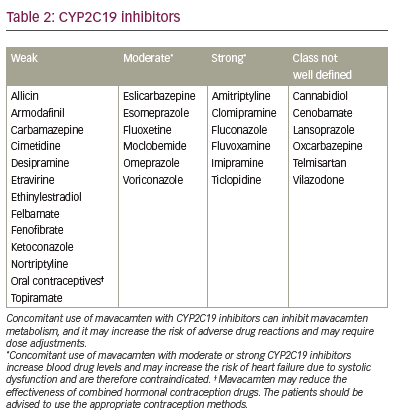
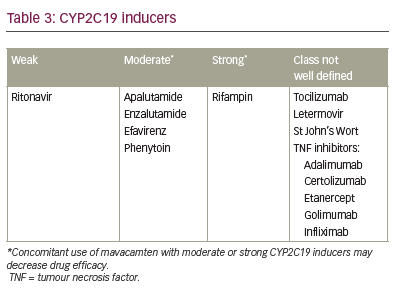
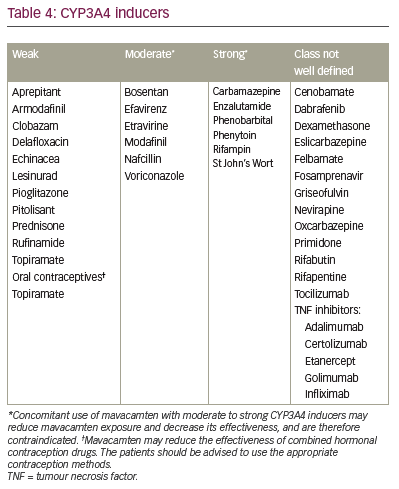
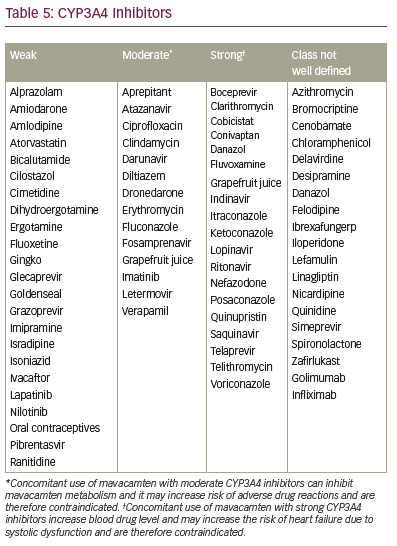
Although mavacamten is a highly effective medication for symptomatic HOCM, there are some difficulties with its use in clinical practice. A limitation to its use in clinical practice is that it should not be used in patients with LVEF <55%, which disqualifies many patients with longer-standing cardiomyopathy. In addition, its use is contraindicated in pregnant patients and patients taking drugs known to interact with the CYP2C19 and CYP3A4 enzymes by either inhibiting or inducing them (Tables 2–5). Regular echocardiographic assessments to evaluate ejection fraction are crucial; however, this adds to the overall cost to the healthcare system and, in some cases, to the patient. Regular medication reconciliations are also key in optimizing the effectiveness and safety of mavacamten.
Conclusions
Until now, the treatment of HOCM had been limited to a few pharmacological agents that have not been studied with optimal scientific rigour and leave with treatment-refractory symptoms a significant proportion of patients who thus require invasive septal reduction therapies to relieve symptoms secondary to LVOT obstruction. Although these techniques are highly effective and, in general, safe when performed in high-volume centres, they do have some residual risk for procedural complications. With the availability of mavacamten, a novel myosin inhibitor targeting the underlying cause of many HCM-related symptoms, the medical treatment of HOCM has become more effective but not trivial in clinical practice. Given the intricacies of mavacamten with the potential for causing reversible systolic heart failure, significant drug–drug interactions and close monitoring requirements, its place in current treatment algorithms has yet to be defined. However, there is great hope and early data suggesting that mavacamten will alter the disease trajectory and thus may soon be the preferred first-line treatment option for patients with HOCM.