Introduction: Long QT syndrome (LQTS) is a rare, heterogenic inherited arrhythmia syndrome. Many affected patients are asymptomatic but LQTS is associated with an increased risk of sudden cardiac death. Decision making around risk management, particularly implantable cardioverter defibrillator (ICD) therapy can be difficult as despite their life saving properties, potential for device related complications and inappropriate shocks is significant, particularly in the younger population.
Aims: This study describes the cohort of genetically confirmed LQT patients within Northern Ireland. We reviewed their clinical features, genetic mutations, and the association of both of these with arrhythmia and ICD implantation.
Methods: The Electronic Care Records of consecutive genetically confirmed LQTS patients (and their families) attending the Northern Ireland Inherited Cardiac Conditions service between 2004–18 were retrospectively analysed. We reviewed their demographics alongside clinical and genetic features at presentation and follow up. Records of 634 patients were reviewed, incorporating 136 families.
Results: The Northern Ireland regional Inherited Cardiac Conditions Service identified 634 patients in Northern Ireland with mutations in KCNQ1, KCNH2, KCNE1, KCNE2 and SCN5A. 397 patients (60%) were female and average follow up was 7.5 years.
Over the 14 years, 50 patients received an ICD, an insertion incidence of 8% of the patient cohort. Significant risk factors influencing device insertion included female sex (p=0.07), history of syncope (p=0.0005), KCNH2 (p=0.003) and KCNQ1 (p=0.001) mutations. Longest documented QTc duration was significantly longer in the device group with an average of 499 ± 51 msec compared to 466 ± 34 msec in non-device group (p<0.0005).
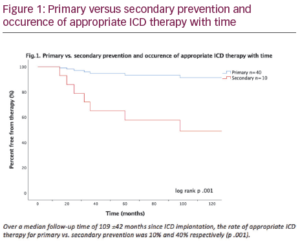
8 patients received appropriate ICD therapy, a shock rate of 1% per year for the patient population. Secondary prevention devices predicted higher therapy rates than primary prevention device (p=0.012) (Figure 1). No other features, including QTc duration, reached statistical significance for prediction of appropriate therapy due to the ICD cohort size. 63.5% (5) of those patients receiving a shock had a KCNH2 mutation compared to 1 patient (12.5%) SCN5A, 1 patient (12.5%) KCNQ1 and 1 patient (12.5%) KCNE1. History of syncope was also suggestive of higher rates of therapy with 87.5% of patients in the therapy group compared to 60% in the
non-therapy group.
Reassuringly, over the 14-year review period there were no deaths in the non-device group. Three patients (6%) received inappropriate shocks with 1 patient in this group also receiving appropriate therapy. There was 1 device associated infection with subsequent device explant.
Conclusion: ICD insertion rates in our region was 8% for genetically confirmed long QT syndrome, a figure significantly lower than many other studies. Factors influencing device decision making were history of syncope, sex and QTc duration. Genetic mutations in KCHN2 and KCNQ1 were also more prevalent in those who received a device. Despite low insertion rates, shock rate was also low at 1% per year. Reassuringly these findings are coupled with no mortality within the non-ICD group, suggesting a non-device approach to management is appropriate in many. Secondary prevention, history of syncope and KNCH2 mutation were more prevalent in those who had appropriate ICD therapy, this information is useful for risk stratification in the future.